
This site is a fork of the original PRABI NPS@ server
|
[HOME]
[DESCRIPTION]
[HELP]
[NEWS]
[CONTACT]
[Geno3D]
July 22, 2024: NPS@ updated (see NEWS).
July 25 17h00 (Paris Time) - July 26 12h00 (Paris Time): NPS@ service interruption.
BLAST help
 A brief introduction to BLAST
A brief introduction to BLAST
BLAST (Basic Local Alignment Search Tool) is the heuristic search algorithm employed by the programs blastp, blastn, blastx, tblastn,
and tblastx; these programs ascribe significance to their findings using the statistical methods of Karlin and Altschul (1990, 1993)
with a few enhancements. The BLAST programs were tailored for sequence similarity searching -- for example to identify homologs
to a query sequence.
For further details, see the NCBI BLAST tutorial.
The programs are :
- blastp : to compare a query protein sequence to a protein sequence databank
- blastx : to compare a query nucleotide sequence (6 frames translation) to a protein sequence databank
- blastn : to compare a query nucleotide sequence to a nucleotide sequence databank
- tblastn : to compare a query protein sequence to a nucleotide sequence databank (6 frames translation)
- tblastx : to compare a query nucleotide sequence (6 frames translation) to a nucleic sequence databank (6 frames translation)
Availability in NPS@
These programs are available :
Parameters
Some parameters are currently not available for the user.
By default the number of description and the number of alignment are set to 500.
The expected threshold is set to 10.0.
The comparison matrix is BLOSUM62.
The default behavior is selected for gap opening (11) and gap extending (1) costs.
To retrieve these informations see the line "Information and statistics : [BLASTP]" in
NPS@ BLAST result file.
NPS@ BLAST output example
The NPS@ BLAST output is divided into three parts.
-
PART 1:
In this part, you have :
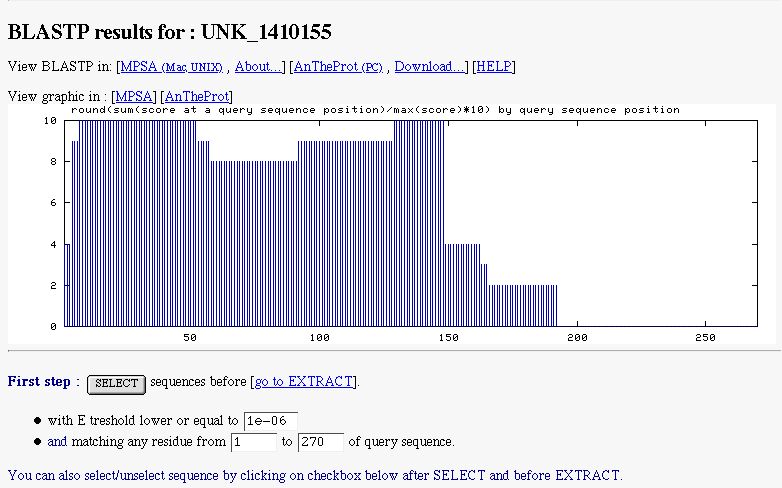
- a graphic indicating the similarity found along the query sequence. It's computed with BLAST pairwise sequence alignment (only for blastp/blastn)
- a form to select subject sequences, you can select sequences by indicated the maximal E-value threshold to do it (by default NPS@ sets it to 1e-6), You can also select subject sequences matching a particular region of the query (this only for blastp/blastn)
-
PART 2:
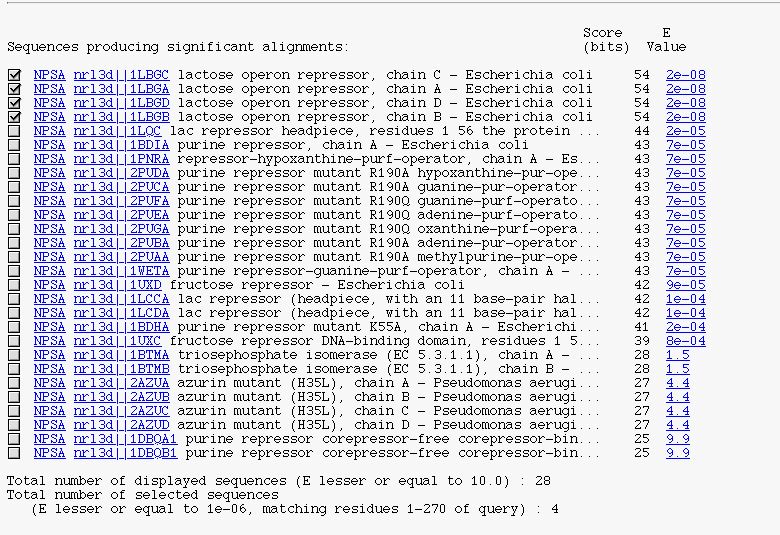
It's the BLAST description block in a 'HTMLized' form.
You can see :
- a checkbox to select/unselect subject sequence
- the NPSA link allows you to apply NPS@ methods on the corresponding sequence after it is extracted from the query database
- the database link to retrieve the database entry
- the alignment link (on expected value) to see BLAST alignment between the query sequence and the current subject sequence
- the number of displayed sequences
- the number of selected sequences
-
PART 3:
You have :
- an extract form to extract selected sequence and make a database. You can extract full or partial (only for blastp/blastn) sequences. The extraction is done on your current selection.
The full extract is made from the database.
The partial extract, is made for subject sequence that match the query one in the indicated region. The extracted sequence is retrieved from the BLAST alignment. You can set the minimal length of the extracted sequence
- a checkbox to add your query sequence in the created database
- a checkbox to remove identical sequences in the created database
- a link on BLAST information and statistics
- a link on the original BLAST result text file

References
Last modification time : Mon Dec 5 15:44:40 2022. Current time : Sat Jul 27 10:13:43 2024. User : public@3.145.63.7.