
This site is a fork of the original PRABI NPS@ server
|
[HOME]
[DESCRIPTION]
[HELP]
[NEWS]
[CONTACT]
[Geno3D]
July 30, 2024: NPS@ updated (see NEWS).
KALIGN help
 A brief introduction to KALIGN
A brief introduction to KALIGN
Kalign is a progressive alignment method striking a good balance between accuracy and speed compared with other alignment programs on a range of popular benchmark datasets.
Kalign program uses the unweighted pair group method with arithmetic mean (UPGMA) algorithm to construct a guide tree resulting in quadratic time complexity.
More recent alignment programs have overcome this hurdle by implementing heuristics to construct guide trees.
Kalign 3 is a completely re-written and updated version to meet current and future alignment challenges, introducing a SIMD (single instruction, multiple data) accelerated version of Gene Myers’ bit-parallel algorithm (Myers, 1999) to estimate pairwise sequence distances and adopting the sequence embedding strategy introduced by Blackshields et al. (2010) to speed up the construction of guide trees.
Availability in NPS@
KALIGN is available :
The set to align cannot have more than 1,000,000 characters.
So, for example, you can use KALIGN to align a set of similar sequences built from a BLAST search.
Parameters
The -super5 option is used for computation.
The output format is CLUSTAL W.
NPS@ KALIGN output example
The NPS@ KALIGN output is divided into three parts.
-
PART 1:
After the reference, you have buttons to post-process the multiple alignment with other software like HMMBUILD, Repertoire, ...
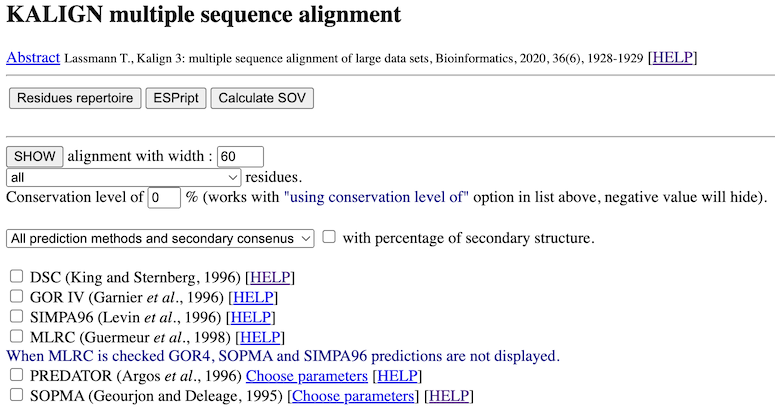
In this part, you have also a form to work with the alignment. Your choices are validate when you click on the SHOW button. You can :
- show residues with different options (identical(*), strongly similar (:), weakly similar (.), ...). The most
interesting option is the "using conservation level" one. With it you, display only residues that have a conservation level
equal or above the value you type in.
- select secondary structure prediction methods to compute and display. This in the case of proteins and when you have no
more than 50 sequences. A method with the "(Do it alone)" sentence has to be computed alone (it must be
the only one newly selected before the next click on "SHOW" button). Otherwise, you won't have your response because of the
timeout (these methods can take 5 minutes or more by sequence). You can also show all methods with or without
secondary consensus or only the consensus. You can
also see the percentage of each secondary structure element (HETC...) for each method.
-
PART 2:
It's the color coded alignment with or without secondary structure predictions inserted.

-
PART 3:
You have :
- The percentage of each secondary structure element (if wanted) for each prediction and for each sequence.
- Some data on the alignment (length, number of identities,...).
- KALIGN options used.
- Links on result text files (KALIGN, secondary structure prediction method outputs,...).
- Secondary structure prediction method parameters choice.

References
Last modification time : Fri Dec 8 17:21:36 2023. Current time : Sun Apr 6 04:28:19 2025. User : public@13.59.69.96.